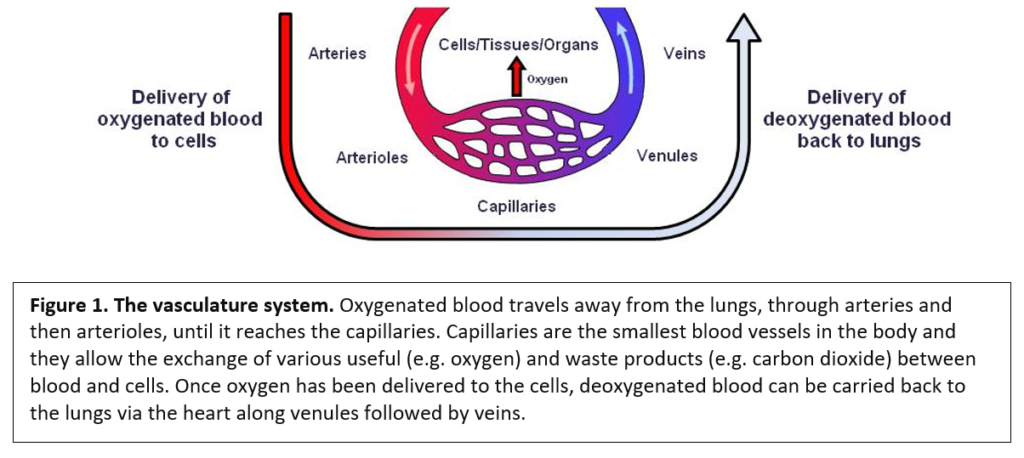
Ilaria Signoria (University Medical Centre, Utrecht, Netherlands), a PhD student working with Ludo Van Der Pol and Ewout Groen, presented her work aiming to find out why there is a wide range of responses in the clinic to the SMN-dependent treatments of nusinersen and risdiplam.
On a population level, the more copies of SMN2 a person has, the less severe the SMA is likely to be. However, on an individual level, this is not always true; for example, people with SMA Type 2 may have two, three or four copies of SMN2. Similarly, when SMN-dependent treatments (e.g. nusinersen and risdiplam) are given to people with the same SMA Type and number of SMN2 gene copies, the effect on symptoms can differ between recipients.
Unfortunately, we do not currently know the reason for these difference between SMA subtypes and between treatment responses.
To provide some insight into this, Ms. Signoria grew skin cells that had been collected from 35 people with SMA (Types 1 to 4) and ten unaffected controls. Each SMA skin sample was taken from people with known SMN genetics and a detailed history of their condition. The skin cells, known as ‘fibroblasts’, are easily obtained, grow and multiply very quickly, and have the same genetic makeup as that of the donor; hence, they are a useful tool for assessing the impact of SMN-dependent therapies in the laboratory.
The skin cells from all 45 people were separately grown and assessed. The shape and size of all cells was evaluated, as were the levels of SMN protein. In addition, the intermediate molecule that is created from DNA to make a protein, known as ‘messenger RNA’, was also assessed. For more information on this, click here.
There were no clear differences in the shape and size of the skin cells from people with SMA compared to controls. However, as expected, there were reductions in the SMN protein and the ‘messenger RNA’ from the SMN1 and SMN2 genes in the SMA ‘fibroblasts’.
Skin cells taken from people with more SMN2 copies generally had higher availability of SMN protein and ‘messenger RNA’. Furthermore, those cells with more SMN2 copies tended to produce more SMN protein when treated with the SMN2-targeting therapies nusinersen and risdiplam.
Interestingly, it was found that the levels of SMN ‘messenger RNA’ did not correlate well with the amount of SMN protein, suggesting that factors involved in converting the ‘messenger RNA’ into protein differ between individuals, and that the process can be more efficient in some people than others.
It was also observed that factors such as age, SMN2 copy number and SMN levels before treatment were only able to explain a proportion of the variability in response to the treatments. This suggests that other currently unknown factors are at play.
The ‘fibroblast’ model is a powerful tool to identify what these factors are. This is exciting because, in the future, we may be able to manipulate these factors in people with SMA to improve individual responses to the SMN-dependent therapies.