SMN Protein: Not Just Important for the Nervous System
SMN Protein: Not Just Important for the Nervous System
Page last updated: 15th November 2011
SMA is principally a disease of lower motor neurons, but observations in type I SMA patients and recent scientific research using mouse models of the disease suggest that the neurons may not be the only cell/tissue affected by reduced levels of the survival motor neuron (SMN) protein. Complications with the heart, brain, bones, and vascular system have all been reported in more severe forms of the disorder. Thomas Gillingwater from the University of Edinburgh gave a nice overview of the contribution of non-motor neuronal cells and tissues in a talk at the 8th annual UK SMA Research Conference.
At first glance it may seem unimportant to know exactly which cells and tissues are affected in the disease. However, a good understanding of which parts of the body deteriorate is in fact vital to identify which tissues in particular need to be targeted by future therapies. For instance, it is no good restoring SMN levels in the lower motor neurons alone if cell and tissue death is occurring independently elsewhere.
A recent scientific paper published in the journal Nature by the Krainer group in Cold Spring Harbor, NY, USA has added to the evidence suggesting that SMN protein will likely have to be restored to most, if not all, tissues for the best chances of successfully treating SMA.
In the study, they used a special molecule called an antisense oligonucleotide in order to increase the amount of SMN protein made by the SMN2 gene in severely affected SMA mice. Humans possess two genes that produce SMN protein – SMN1 and SMN2. Fully functional, normal SMN protein is made from the SMN1 gene, and this is the gene that, when mutated, leads to SMA. Contrastingly, everyone’s SMN2 gene only produces about 10% of the working protein that SMN1 in unaffected individuals makes. This is because SMN2 has a few small changes in its genetic code (the DNA sequence), causing it to make less functional SMN protein. Antisense oligonucleotides are small molecules that can be designed to bind very specifically to pre-determined stretches of DNA and mRNA. The antisense oligonucleotide used in the study, called ASO-10-27, is able to target and bind to the important region of mRNA that differs between SMN1 and SMN2, encouraging the production of more working SMN protein from the SMN2 gene (see figure below).
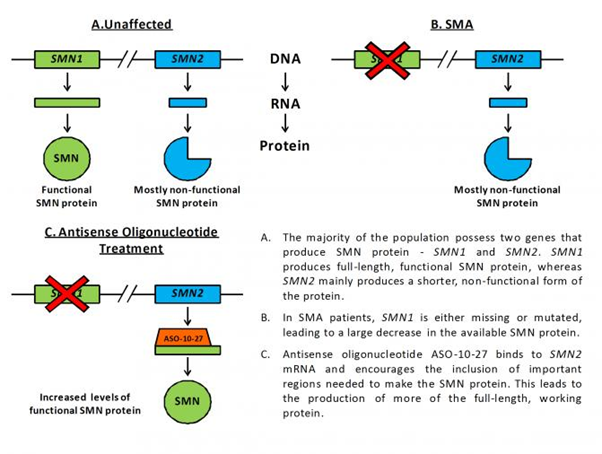
Injection of ASO-10-27 into the brains of newborn SMA mice caused a striking increase of SMN protein in the nervous system, yet resulted in only a marginal improvement in lifespan of a few days. This result suggests that increasing SMN levels in the nervous system alone may not be sufficient for treating disease.
In stark contrast, systemic therapy delivery via injection into the skin, which elevated SMN levels throughout the body during the first few weeks of life, caused a drastic increase in lifespan from approximately two weeks to over 100 days. Furthermore, a combination of the two types of injections resulted in an even greater improvement, as did delivery of greater quantities of the drug and multiple injections.
Importantly, systemic ASO-10-27 treatment also improved disease symptoms in the SMA mouse, causing a reduction in lower motor neuron death and neuromuscular junction pathology, and an improvement in heart weight, and muscle size and strength.
Together, these experiments in the SMA mouse suggest that SMN protein levels will need to be restored throughout the body, and not just the nervous system, in order to have the best chances of successfully treating the disease in patients.