Azzouz Laboratory: Gene Therapy for SMA
Azzouz Laboratory: Gene Therapy for SMA
Page last updated: 12th February 2013
SMA is caused by low levels of an essential protein called the survival motor neuron (SMN) protein. The function of SMN is so important that just about every cell in the body has some SMN.
SMN is made from two very similar genes called SMN1 and SMN2 (see figure). The SMN1 gene produces all the SMN protein we need to survive and live a healthy life. SMN2 on the other hand makes only about 10% of that. Patients with SMA lack a working SMN1 gene, which means that the amount of available SMN protein is drastically reduced. This leads to the death of lower motor nerves, which connect the brain to muscles allowing voluntary muscle contraction, and the development of SMA.
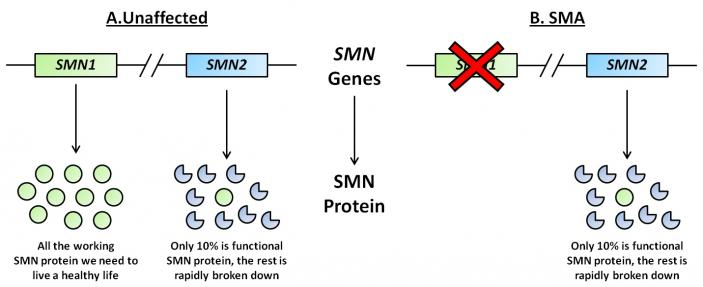
There are differing severities of SMA, and this is because people lacking SMN1 have varying numbers of the SMN2 gene. Someone with four copies of SMN2 is able to produce more SMN protein than someone with only two copies. The more SMN protein a patient with SMA has, the less severe on average their condition will be.
This knowledge that the severity of SMA is heavily dependent on the availability of SMN has caused scientists to think up ways of putting SMN protein back into SMA patients, in the hope that this will cure or at least improve the disease.
That may sound like a relatively easy thing to do; we inject medicines into the body all the time, just think of vaccinations, or insulin for diabetics. Unfortunately, it is not that simple. The injected SMN would need to find its way to the lower motor nerves affected by the disease. Not only that, in order to not require many thousands of injections, it would have to be able to spread to other nerves by itself, and remain biologically active for long periods of time. Also, research suggests that SMN would need to be restored in other cells and tissues besides the lower motor nerves to cure the more severe forms of the disease.
This is where research co-funded by the Jennifer Trust (now SMA UK) from the laboratory of Prof. Mimoun Azzouz comes in.
Using Viruses To Treat SMA
The Azzouz group have been developing a new SMA gene therapy that makes use of viruses to replace the SMN protein lost by SMN1 gene mutations. The principle is that SMN1 is packaged into harmless viruses, which can then be injected into SMA patients. These viruses are then able to travel around the body, targeting various cells including the motor neurons, where they produce SMN protein for long periods of time.
First publishing in 2004, Prof. Azzouz is one of the pioneers of developing and testing virus-based gene therapies for SMA. In this work from 2004, injection of SMN protein viruses into a mouse model of SMA increased SMN protein levels, reduced motor neuron death, and resulted in a modest improvement in lifespan (about a 30% extension compared to untreated mice).
Much has been learnt since Prof. Azzouz’s first SMA publication. Using viruses to replace SMN protein in SMA patients is now considered to be one of the most promising potential treatments for the disease. This is in part due to the Jennifer Trust-funded work by Prof. Azzouz and colleagues published in 2010 in the well-respected scientific journal Science Translational Medicine (Vol 2, Issue 35, ra42).
In this study, one day old SMA mice were given a single intravenous (into a vein) injection of viruses able to produce SMN protein.
The viruses were shown to reach all levels of the spinal cord, successfully targeting and producing SMN protein in approximately 55% of the motor neurons. SMN levels were also increased in muscle and the liver.
This restoration of SMN protein in the SMA mice resulted in improvements in motor function, body weight gain, and most importantly lifespan. 8 out of 10 mice receiving the treatment showed an increase in survival, with each mouse living on average about six times longer than untreated controls.
Unfortunately, the SMN virus-treated mice did not reach the natural life expectancy of healthy mice (>2 years), but died due to various complications including respiratory distress. This is very likely due to the incomplete restoration of SMN protein levels in all of the necessary cells, rather than a toxic effect of the treatment, because healthy control mice treated with the same viruses showed no such effect.
Nevertheless, this improved survival of SMA mice caused by SMN gene replacement is amongst the best increases in lifespan reported to date.
We are a few years away from translating this work in SMA mice to clinical trials in SMA patients, and we must be cautious because the treatment may not be as effective in humans due to, for example, differences in our immune response that renders the viruses impotent.
That being said, Prof. Azzouz’s group and other laboratories from around the world are really driving forward viral gene therapy as a viable option for potentially treating and hopefully curing SMA in the near future.