Spinal Muscular Atrophy (SMA) – A Brief Summary
Spinal Muscular Atrophy (SMA) – A Brief Summary
Who this is for
This information is for anyone who wants a brief summary of the impact of SMA. It covers what causes the condition, the numbers of people affected and possible treatments.
Spinal Muscular Atrophy is a rare, neuromuscular condition. It causes progressive muscle wasting (atrophy) and weakness. It may affect crawling and walking ability, arm, hand, head and neck movement, breathing and swallowing. How severely people are affected, and in what way, varies greatly.
Most people have two genes called the Survival Motor Neuron 1 (SMN1) gene. This produces Survival Motor Neuron (SMN) protein which keeps lower motor neuron nerve cells healthy.
These nerve cells are essential for activating muscles used for crawling and walking, the movement of arms, hands, head and neck, as well as breathing and swallowing.
Most people have two healthy copies of the SMN1 gene. People who have 5q SMA have two ‘altered’ copies of the SMN1 gene. This means they are unable to produce enough SMN protein to have healthy lower motor neurons.
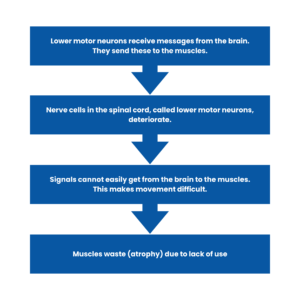
As a result, the lower motor neurons in the spinal cord deteriorate.
This means that signals are not effectively carried from the brain to the muscles. This makes movement difficult.
The muscles then waste due to a lack of use — this is known as muscular atrophy.
In summary:
A second gene also produces SMN protein. This is the SMN2 gene. It is sometimes referred to as the SMA ‘back-up’ gene.
Only some of the SMN protein the SMN2 gene makes works properly. The SMN2 gene cannot fully make up for the altered SMN1 genes in people who have 5q SMA.
People can have between 0 – 8 copies of the SMN2 gene (SMN2 copy numbers). Having more SMN2 copies is generally associated with less severe SMA symptoms. However, a person’s SMN2 copy number does not fully predict their SMA Type or severity.
Before the new drugs for 5q SMA were developed, clinicians studied the effects of SMA on people. This is called the ‘natural history’ of the condition.
This led to 5q SMA being divided into four main Types of SMA: Types 1, 2, 3, and 4. Sometimes a baby is affected before birth; this is called Type 0.
These ‘Types’ of SMA are based on the age that symptoms begin, and what physical milestones (e.g. sitting, standing, walking) are achieved.
These clinical classifications are still used by doctors for adults, teenagers and children living with SMA. For many, though, care and treatment are changing their SMA outcomes.
| SMA Type | Age symptoms usually begin | Motor milestones |
|---|---|---|
| Type 1 | 0-6 months | Unable to sit or roll independently |
| Type 2 | 7-18 months | Able to sit but not walk independently |
| Type 3 | 18 months – 18 years | Able to walk though may lose this ability over time |
| Type 4 | 18 years + | Mild walking difficulties |
How severe and what impact SMA has varies from person to person, both within and between ‘Types’. Each child and adult is affected differently.
-
 Every month in the UK, 4 babies are born with 5q SMA.
Every month in the UK, 4 babies are born with 5q SMA.
5q SMA affects an estimated 1 in 14,000 births. In 2023, this would mean that:
- around 47 babies in the UK were born with 5q SMA.
- approximately 28 of these babies (60%) would have the more severe SMA Type 1.
There is no current evidence to confirm this incidence for the UK population. The SMA Newborn Screening Inservice Evaluation (ISE) > is using 1 in 10,000 as an estimated incidence.
This suggests that in 2023:
- approximately 66 babies were born who will develop a type of 5q SMA
- approximately 40 of these babies (60%) would have the more severe SMA Type 1.
Further UK population research will allow a more accurate measure of SMA incidence in the UK.
-
 In 2023 there were up to an estimated 1,365 people living with 5q SMA in the UK.
In 2023 there were up to an estimated 1,365 people living with 5q SMA in the UK.
As there is no central source of information, exact numbers are unknown. Worldwide, between 1 and 2 people in every 100,000 have 5q SMA.
-
 Although SMA is a rare condition, an estimated 1 in 40 people carry the altered gene. That’s around 1.7 million people in the UK.
Although SMA is a rare condition, an estimated 1 in 40 people carry the altered gene. That’s around 1.7 million people in the UK.
When two ‘carriers’ of the altered gene have a child, for each and every pregnancy there is a one in four chance that the child will have SMA.
There is no cure for SMA and until late 2019 there were no NHS-approved drug treatments specifically for SMA in the UK. There are now three NHS-funded drug treatments:
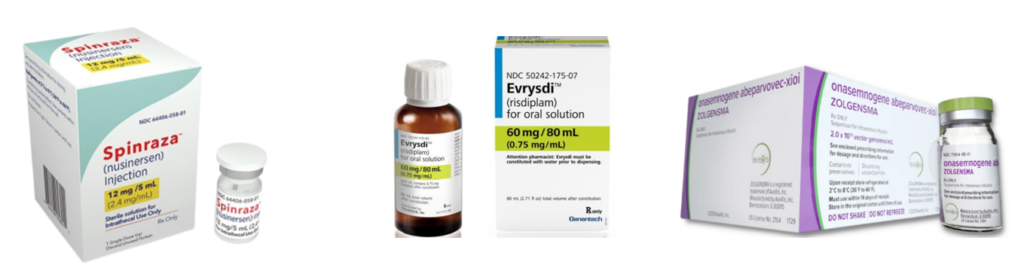
These treatments are not suitable for everyone who has 5q SMA, but most people who have SMA Type 1, 2 or 3 can receive one of them.
These drugs and better care and management of the condition can change what motor milestones (e.g. the ability to sit, stand and walk) babies and children may be able to achieve, and improve their general health.
The treatments work best if started before there is any muscle weakness, or when this is minimal. It is therefore important for treatment to be started as soon as possible.
This is why clinicians and patient groups are calling for the earliest possible introduction of Newborn Screening for SMA in the UK.
For adults living with SMA, drug treatment that can stabilise the condition later in life may also make a positive difference – for example, helping with fatigue or preventing the loss of the ability to use a finger to control a powerchair or laptop.
Much more is known about 5q SMA than the other very rare forms of SMA which have different genetic causes and inheritance patterns. For information about these conditions, including what is known about their symptoms and causes, please see Rarer Forms of SMA.
Was this page useful?
![]() Version 7
Version 7
Author: SMA UK Information Production Team
Last reviewed: November 2024
Next full review due: November 2025
Links last checked: November 2024
This page, and its links, provide information. This is meant to support, not replace, clinical and professional care.
Find out more about how we produce our information.