What is 5q Spinal Muscular Atrophy?
What is 5q Spinal Muscular Atrophy?
Spinal Muscular Atrophy (SMA) is a rare, neuromuscular condition. It causes progressive muscle wasting (atrophy) and weakness. It may affect crawling and walking ability, arm, hand, head and neck movement, breathing and swallowing.
There are several forms of SMA that have different genetic causes and a wide spectrum of how severely children and adults are affected. The most common form is known as ‘5q SMA’, which includes SMA Types 1, 2, 3 and 4. The term ‘5q’ refers to its genetic cause.
For information about some of the rarer forms of SMA that have different genetic causes (non-5q SMA), see: Rarer Forms of SMA >.
-
The SMN1 gene1
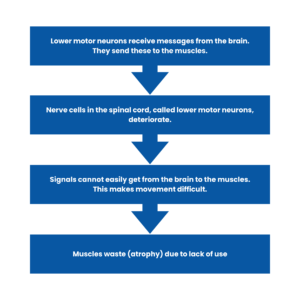
5q SMA affects the nerve cells called lower motor neurons.
Lower motor neurons are found within the spinal cord. They receive electrical signals from the brain and transfer these signals to the muscles This leads to movement – such as crawling and walking.
In the same way, the lower motor neurons also carry signals to the muscles we use for breathing and swallowing.
For these nerve cells to be healthy, our Survival Motor Neuron 1 (SMN1) genes1 must make enough Survival Motor Neuron (SMN) protein.
The SMN1 gene is on chromosome 5 in the region labelled ‘q’. This is why this form of SMA is called ‘5q SMA’.
For more information, see: The Genetics of 5q SMA >.
Most people have two copies of the SMN1 gene. People with 5q SMA have two altered copies of the SMN1 gene. They are unable to produce enough working (functional) SMN protein to have healthy lower motor neurons2.
As a result, the lower motor neurons in the spinal cord deteriorate.
This means that signals are not effectively carried from the brain to the muscles. This makes movement difficult.
The muscles then waste due to a lack of use — this is known as muscular atrophy.
In summary:
- The SMN2 gene1
A second gene is also able to make SMN protein. This is the Survival Motor Neuron 2 gene (SMN2). It is sometimes called the SMA ‘back-up’ gene.
However, researchers have shown that only about 1 in 10 (10%) of the SMN protein made from SMN2 works properly. The remaining 9 out of 10 (90%) of the protein made from SMN2 is missing an essential part — so, it does not work3.
So, although SMN2 can make some functional SMN protein, it cannot produce enough to fully make up for the altered SMN1 gene in people with 5q SMA.
Unlike most genes, the number of copies of SMN2 on each chromosome can vary from one person to the next4. There can be between 0 and 8 copies.
Looking across all people with 5q SMA, the severity of SMA is linked to SMN protein levels4,5. There is a general relationship between the number of SMN2 copies and the likely severity of SMA symptoms.
Having more SMN2 copies is generally associated with less severe SMA symptoms. However, a person’s ‘SMN2 copy number’ does not fully predict their SMA type or severity6,7. This is likely to be because other genetic, and possibly environmental, factors have an influence on the condition.
Bearing this in mind, the following table gives a summary of:
- how many SMN2 copies the majority of people with each Type of SMA may have (see ‘usual number’ column), and
- the possible range of copy numbers that people with each Type of SMA can have (see ‘range’ column)

Before the new drugs for 5q SMA were developed, clinicians studied the effects of SMA on people. This is called the ‘natural history’ of the condition.
This led to 5q SMA being divided into four main Types of SMA: Types 1, 2, 3, and 4. Sometimes a baby is affected before birth; this is called Type 0.
These ‘Types’ of SMA are based on the age that symptoms begin, and what physical milestones (e.g. sitting, standing, walking) are achieved. Clinicians agreed that there could be variation both within and between ‘Types’.
| SMA Type | Age symptoms usually begin | Motor milestones |
|---|---|---|
| Type 1 | 0-6 months | Unable to sit or roll independently |
| Type 2 | 7-18 months | Able to sit but not walk independently |
| Type 3 | 18 months – 18 years | Able to walk though may lose this ability over time |
| Type 4 | 18 years + | Mild walking difficulties |
*Adapted from Finkel et al., 20179
This clinical classification system is still used in the UK for adults, teenagers and children living with SMA. For many, though, care and treatment are changing their SMA outcomes.
-
Due to symptoms of SMA
SMA is confirmed via a test for the SMN1 gene. The classification system of ‘Type’ is used when a baby, child, young person or adult shows symptoms.
Parents / carers will often have been concerned about symptoms of weakness in their child. Symptoms may have been noticed by doctors, the health visitor or community nurse.
Adults who are diagnosed with SMA Type 4 may have had concerns about their muscle weakness or fatigue.
A GP may have met few, or even no children or adults who have SMA. They should make an immediate referral to a Regional (Specialist) Neuromuscular Centre >.
A paediatrician or neurologist will then examine any child or adult with suspected SMA. They will also ask about their medical history and any concerns.
A blood sample is then taken for DNA (genetic) testing. This looks for a ‘deletion mutation’ in the Survival Motor Neuron 1 (SMN1) gene on chromosome 5.
The result of the SMN1 deletion test is usually available within 2 weeks though sometimes it can take longer.
At the same time, the number of copies of the SMN2 gene (the back-up gene for SMN1) is also tested. Having more copies of the SMN2 gene is generally associated with less severe symptoms of SMA.
-
Due to newborn screening:
There is currently a newborn screening research study based in the Oxford – Thames Valley – Wessex region > that parents may choose to join.
Any baby diagnosed with SMA will have had a newborn screening test that looks for the SMN1 gene deletion and the number of copies of the SMN2 gene. The number of SMN2 copies indicates what symptoms and effects the baby’s SMA would be likely to cause if they do not receive drug treatment and appropriate care. The baby may or may not be showing symptoms at this stage.
The Generation Research Study > that parents may choose to join also includes screening for SMA.
The UK National Screening Committee (NSC) is responsible for assessing the evidence for national screening programmes and recommending which conditions should be screened for.
The NSC is working on developing an In-Service Evaluation (ISE) of newborn screening for SMA in the UK >.
There is no cure for SMA and until late 2019 there were no NHS-approved drug treatments specifically for SMA in the UK.
There are now three NHS-funded, disease-modifying drug treatments:
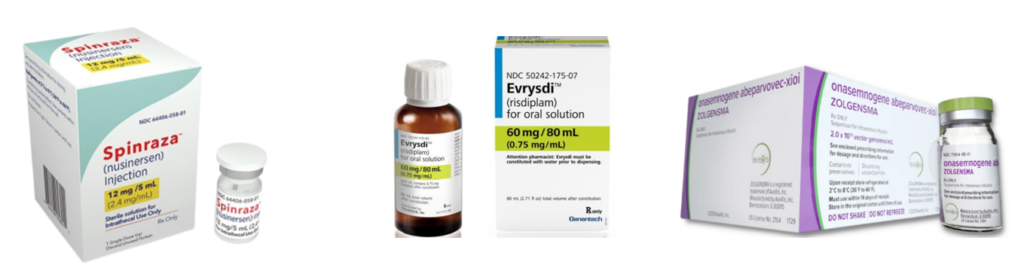
These treatments are not suitable for everyone who has 5q SMA, but most people in the UK who have SMA Type 1, 2 or 3 can receive one of them. There have been no clinical trials of any of the treatments with people who have SMA Type 4.
These drugs can change what motor milestones babies and children may be able to achieve, and improve their general health.
The treatments work best if started before there is any muscle weakness, or when this is minimal. It is therefore important for treatment to be started as soon as possible10.
This is why clinicians and patient groups are calling for the earliest possible introduction of newborn screening for SMA in the UK. You can read about progress towards this on our page on Newborn Screening for SMA >
For adults living with SMA, drug treatment that can stabilise the condition later in life may also make a positive difference – for example, helping with fatigue or preventing the loss of the ability to use a finger to control a powerchair or laptop.
For summaries about these NHS-funded drug treatments, see:
Whatever the outcome, any of these drug treatments must be combined with the best supportive care and management of symptoms.
The International Standards of Care for SMA’ (SoC)7,11 were agreed in 2017. They were summarised the following year in the Family Guide >.
The recommended standards of care for children, young people and adults varied and were based on:
- whether they could sit, stand or walk
- whether their breathing was affected by their SMA
- what other daily living activities they could manage.
Though they are still important, these standards were written before the new disease-modifying drug treatments became more widely available.
This is why a 3-year project is now underway to update these standards for the UK. Clinicians and patient reps are reviewing all aspects of care and management.
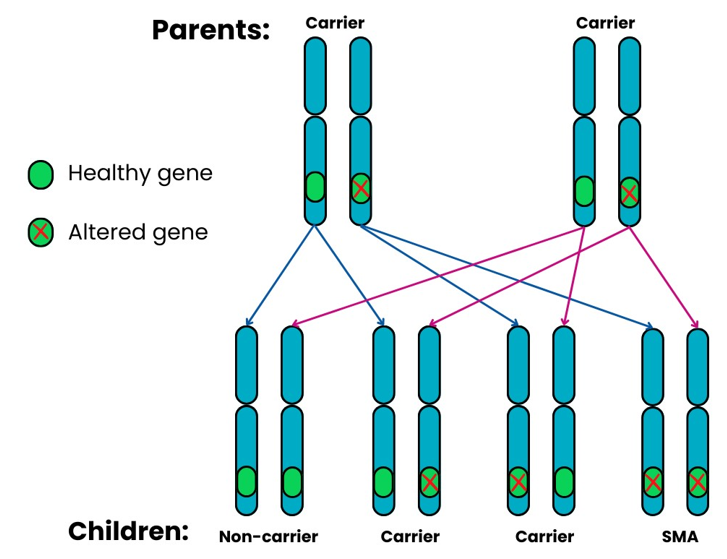
5q SMA is passed from parents to their children through altered SMN1 genes. This usually follows an autosomal, recessive pattern of inheritance. It means that:
When two SMA carriers have a child together, for each pregnancy there is a:
- 1 in 4 (25%) chance that the child will inherit both altered copies of the SMN1 gene and will have SMA.
- 2 in 4 (50%) chance that the child will inherit one altered copy and one healthy copy of the SMN1 gene and will be a carrier.
- 1 in 4 (25%) chance that the child will inherit two healthy copies of the SMN1 gene and will not be a carrier or have SMA.
A very small number of people develop 5q SMA despite standard carrier tests showing that one or both parents are not carriers. For example, for around 2 in every 100 (2%) people who have 5q SMA, the ‘alteration’ (when part of the DNA changes or is missing) has occurred for the first time.
It is important that all parents have genetic counselling, including anyone who is a ‘non-carrier’. Genetic counselling explores each person’s specific circumstances.
For more detailed information, please see: The Genetics of 5q SMA >.
-
 An estimated 1 in 40 people carry the altered SMN1 gene12.
An estimated 1 in 40 people carry the altered SMN1 gene12.
This means that although SMA is a rare condition, around 1.7 million people13 in the UK are carriers of the altered SMN1 gene.

-
Every month in the UK, 4 babies are born with 5q SMA.
This is the incidence of SMA – the number of new people diagnosed with SMA at any one time.
Pooling all published newborn screening data has shown that approximately one in every 14,000 babies worldwide are born with a Type of 5q-linked SMA14,15.
For every 100 who have SMA, approximately 60 babies would have the most severe SMA Type 1 (60%)16.
In the UK in 2023, there were 664,400 live births 13. This suggests that:
- approximately 47 babies were born who will develop a Type of 5q SMA.
- approximately 28 of these babies (60%) would have the more severe SMA Type 1.
There is no current evidence to confirm this incidence for the UK population. The SMA Newborn Screening Inservice Evaluation (ISE) > is using 1 in 10,000 as an estimated incidence. This suggests that in 2023:
- approximately 66 babies were born who will develop a type of 5q SMA.
- approximately 40 of these babies (60%) would have the more severe SMA Type 1.
Further UK population research will allow a more accurate measure of SMA incidence in the UK.
-
Global studies suggest that worldwide, between 1 and 2 people in every 100,000 have a Type of 5q SMA12.
 This is the prevalence – how many people are living with SMA in a population at any one time.
This is the prevalence – how many people are living with SMA in a population at any one time.
In 2023, the UK population was approximately 68.3 million13. The global studies therefore suggest that:
- between 683 and 1,366 people have SMA in the UK at any one time.
There is no central information source, so the exact number is unknown.
These global studies were published before drug treatments became available. These treatments have had a life-saving impact for many children who have SMA Type 1. They have led to life-changing, healthier lives for many others.
It therefore seems reasonable to suggest that the prevalence of SMA in the population will slowly increase year by year.
There is still a considerable amount of research into SMA taking place around the world, and there are more drugs being tested in clinical trials. This will continue to improve our understanding of SMA and the effects of, and responses people have to, the current treatments. It will also help with the further development of effective management and care.
To keep up-to-date with the latest developments in research and drug treatments, see Treatments and Research >.
1. Lefebvre S et al. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80: 155-165.
2. Lunn MR & Wang CH (2008) Spinal muscular atrophy. Lancet 371: 2120-2133.
3. Ruggiu M et al. (2012) A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol 32: 126-138.
4. Mailman MD et al. (2002). Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med 4: 20-26.
5. Lefebvre S et al. (1997). Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16: 265-269.
6. A Guide to the 2017 International Standards of Care for SMA. (Last accessed: 21st October 2024).
7. Mercuri E et al. (2018) Diagnosis and management of spinal muscular atrophy: Part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 28: 103-115.
8. Tillmann R et al. (2018) Spinal Muscular Atrophy (SMA) type 1, a changing phenotype: implications for motor function and physiotherapy management from the Nusinersen Expanded Access Program (EAP). APCP Journal 9: 4-12.
9. Finkel RS et al. (2017) ENMC SMA Workshop Study Group. 218th ENMC International Workshop: Revisiting the consensus on standards of care in SMA Naarden, The Netherlands, 19-21 February 2016. Neuromuscul Disord 27: 596-605.
10. Dangouloff T & Servais L (2019) Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther Clin Risk Manag 15: 1153-1161.
11. Finkel RS et al. (2018) Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord 28: 197-207.
12. Verhaart IEC et al. (2017) Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis 12: 124.
13. Population estimates for the UK, England, Wales, Scotland and Northern Ireland: mid-2023 – published 8th October 2024 (last accessed 21st October 2024)
14. Aragon-Gawinska et al. (2023) Spinal muscular atrophy treatment in patients identified by newborn screening—A systematic review. Genes 14: 1377.
15. Belter et al (2024) Newborn Screening and Birth Prevalence for Spinal Muscular Atrophy in the US” in JAMA Pediatrics on July 15, 2024.
16. Verhaart IEC, et al. (2017) A multi-source approach to determine SMA incidence and research ready population. J Neurol 264: 1465-1473.
Was this page useful?
![]() Version 10
Version 10
Author: SMA UK Information Production Team
Last updated: November 2024
Next full review due: November 2025
Links last checked: November 2024
This page, and its links, provide information. This is meant to support, not replace, clinical and professional care.
Find out more about how we produce our information.